Successful modern drug discovery research makes extensive use of structural data on protein drug targets, candidate drug molecules, and complexes of the two. However, rationally analysing the wealth of structural data in a scalable manner to drive the discovery of new active pharmaceutical ingredients (APIs) can prove difficult. CSD-CrossMiner leverages the well-known concept of a pharmacophore to do just that—empowering researchers to discover important new therapeutics.
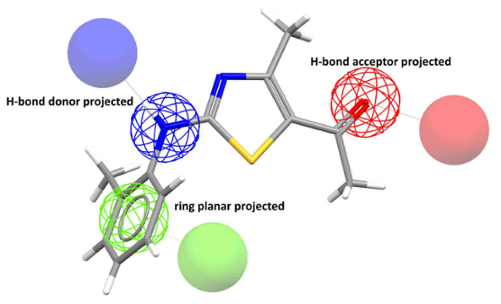
Example of a pharmacophore model. The green sphere represents a ring
feature, the red sphere represents a hydrogen-bond acceptor feature, and the blue sphere represents a hydrogen-bond donor feature.
A pharmacophore is a model molecule with an ensemble of the necessary steric and electronic features to ensure interactions with a target structure. With CSD-CrossMiner, researchers can mine world-leading databases—like the Cambridge Structural Database (CSD) and the Protein Data Bank (PDB)—and proprietary datasets with a pharmacophore-based screening of general chemical features, like the presence of rings, hydrogen bonding, hydrophobicity, and more.
Download this free white paper to learn how adding CSD-CrossMiner to your drug discovery workflow and virtual screenings can:
- Lead to scaffold-hopping: identify new scaffolds that can inform the design of molecular mimics of established lead compounds and find new APIs for established targets.
- Find alternative fragments, bioisosteres, and similar pockets: cohesively analyse your target for all available chemistry.
- Empower medicinal chemists to find the most pharmaceutically viable solid forms: intuitively analyse cross-pharmacology between protein targets and better understand potential risks around toxicity and adverse reactions.
